Rapid Antigen Testing for SARS-CoV-2
General Guidance
Rapid antigen tests are commonly used in the diagnosis of respiratory pathogens, including influenza viruses and respiratory syncytial virus (RSV). The FDA has granted emergency use authorization (EUA) for antigen tests that can identify SARS-CoV-2. See FDA’s list of In Vitro Diagnostics EUA.
Antigen tests are immunoassays that detect the presence of a specific viral antigen, which implies current viral infection. Antigen tests are currently authorized to be performed on nasopharyngeal or nasal swab specimens placed directly into the assay’s extraction buffer or reagent. The currently authorized antigen tests are not restricted to use on persons of a certain age. See Table 2 for additional information about antigen tests.
Antigen tests are relatively inexpensive and can be used at the point-of-care. The currently authorized devices return results in approximately 15 minutes. Antigen tests for SARS-CoV-2 are generally less sensitive than viral tests that detect nucleic acid using reverse transcription polymerase chain reaction (RT-PCR). See FDA’s list of In Vitro Diagnostics EUA for more information about the performance of specific authorized tests. Proper interpretation of antigen test results is important for accurate clinical management of patients with suspected COVID-19, or for identification of potentially infected persons when used for screening.
The clinical performance of rapid antigen diagnostic tests largely depends on the circumstances in which they are used. Rapid antigen tests perform best when the person is tested in the early stages of infection with SARS-CoV-2 when viral load is generally highest. They also may be informative in diagnostic testing situations in which the person has a known exposure to a confirmed case of COVID-19. Rapid antigen tests can be used for screening testing in high-risk congregate settings in which repeat testing could quickly identify persons with a SARS-CoV-2 infection to inform infection prevention and control measures, thus preventing transmission. In this case, there may be value in providing immediate results with antigen tests even though they may have lower sensitivity than RT-PCR tests, especially in settings where a rapid turnaround time is required. See FDA’s FAQs on screening asymptomatic individuals and use of antigen tests in high risk congregate settings.
There are limited data to guide the use of rapid antigen tests as screening tests on asymptomatic persons to detect or exclude COVID-19, or to determine whether a previously confirmed case is still infectious.
Clinicians should understand antigen test performance characteristics in order to recognize potentially false negative or false positive results and to guide patient management. Laboratory and testing professionals who perform rapid antigen tests should also understand the factors that affect the accuracy of antigen testing, as described in this guidance.
Regulatory Requirements for Using Rapid Antigen Tests for SARS-CoV-2
FDA regulates in vitro diagnostic devices and has provided recommendations and information regarding EUA requests for COVID-19 diagnostic tests in the Policy for Coronavirus Disease-2019 Tests During the Public Health Emergency (Revised) (“Policy for COVID-19 Tests”) and the EUA templates referenced in that policy. COVID-19 assays and test systems used for diagnostic or screening testing, including those for antigen testing, must have received an EUA from FDA or be offered under the policies in FDA’s Policy for COVID-19 Tests. Any rapid antigen test for SARS-CoV-2 authorized for use by FDA will be included on FDA’s list of In Vitro Diagnostics EUA.
Laboratory and testing professionals who conduct diagnostic or screening testing for SARS-CoV-2 with rapid antigen tests must also comply with Clinical Laboratory Improvement Amendments (CLIA) regulations. Any laboratory or testing site that intends to report patient-specific test results must first obtain a CLIA certificate and meet all requirements to perform that testing. For more information, see the Centers for Medicare & Medicaid Service’s (CMS) summary of the CLIA regulations. CMS has provided additional information on enforcement discretion for the use of SARS-CoV-2 point-of-care antigen tests on asymptomatic individuals on its website.
Laboratory and testing professionals who conduct surveillance testing for SARS-CoV-2 with rapid antigen tests are not obligated to comply with these FDA and CLIA requirements. However, CDC recommends that facilities that conduct surveillance testing for SARS-CoV-2 with antigen tests should use an antigen test that has been authorized for use, which are listed on FDA’s In Vitro Diagnostics EUA. Information on surveillance testing performed by Universities may be found on the CMS website.
Collection and Handling of Clinical Specimens
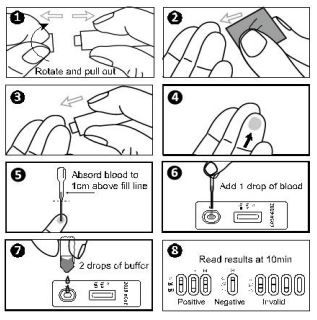
All testing for SARS-CoV-2, including rapid antigen testing, is directly impacted by the integrity of the specimen, which depends on specimen collection and handling. Improper specimen collection may cause some swabs to have limited amounts of viral genetic or antigenic material for detection. Inadequate quality assurance procedures could result in cross contamination of the specimen, which could cause inaccurate test results. Delays from sample collection to testing should be minimized. Biosafety measures and instructions for use should be followed precisely to ensure accurate testing and safety of those who perform the testing. See CDC’s guidance on Collecting, Handling, and Testing Clinical Specimens for COVID-19.
Some antigen assays have explored the use of viral transport media, but the introduction of this dilution may decrease the sensitivity of the assay and carries the risk of cross contamination that can cause false-positive results. Laboratories and testing sites should follow the instructions for use and the package insert that are specific for the test system that they are using.
Performance of Rapid Antigen Tests for SARS-CoV-2
It is important for clinicians and testing personnel to understand the performance characteristics, including analytic sensitivity and specificity, of the particular rapid antigen test being used, and to follow the manufacturer’s instructions and package insert.
The “gold standard” for clinical diagnostic detection of SARS-CoV-2 remains RT-PCR. Thus, it may be necessary to confirm a rapid antigen test result with a nucleic acid test, especially if the result of the antigen test is inconsistent with the clinical context. When confirming an antigen test result with a RT-PCR test, it is important that the time interval between collection of samples for the two tests is less than two days, and there have not been any opportunities for new exposures between them. If more than two days separate the two collections, or if there have been opportunities for new exposures, the nucleic acid test should be considered a separate test – not a confirmatory test. Table 2 summarizes the differences between RT-PCR tests and antigen tests.
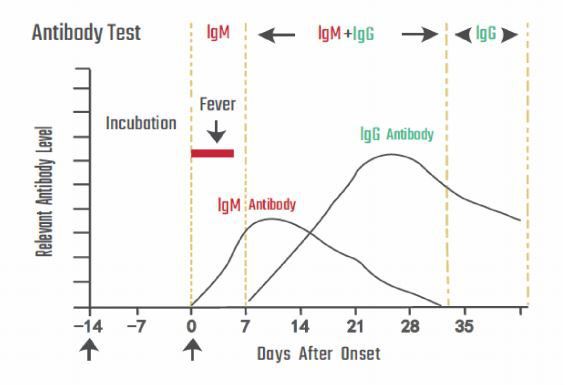
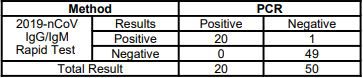
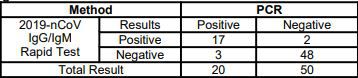
The sensitivity of rapid antigen tests is generally lower than RT-PCR. The first antigen tests to have received FDA EUAs demonstrate sensitivity ranging from 84.0%-97.6% compared to RT-PCR. Antigen levels in specimens collected beyond 5-7 days of the onset of symptoms may drop below the limit of detection of the test. This may result in a negative test result, while a more sensitive test, such as RT-PCR, may return a positive result.
The specificity of rapid antigen tests is generally as high as RT-PCR – the first antigen tests that have received FDA EUAs have reported specificity of 100% – which means that false positive results are unlikely. Positive and negative predictive values of all in vitro diagnostic tests vary depending upon the pretest probability of the patient being tested. Pretest probability is impacted by the prevalence of the target infection in the community as well as the clinical context of the recipient of the test. Table 3 provides additional information on the relationship between pretest probability and the likelihood of positive and negative predictive values.
CDC recommends that laboratory and testing professionals who perform rapid antigen testing should determine infection prevalence based on a rolling average of the positivity rate of their own SARS-CoV-2 testing over the previous 7–10 days. Infection prevalence at the time of testing, as well as the clinical context of the recipient of the test, impacts pretest probability. If a specific testing site, such as a nursing home, has a positivity rate near zero, the prevalence of disease in the community (e.g., cases per population) should instead be used to help determine pretest probability. Rapid antigen tests should be interpreted in the context of the prevalence of infection or disease, the device’s performance characteristics and instructions for use, and the patient’s clinical signs, symptoms, and history.
Evaluating the Results of Rapid Antigen Testing for SARS-CoV-2
Evaluating the results of a rapid antigen test for SARS-CoV-2 should take into account the performance characteristics (e.g. sensitivity, specificity), instructions for use of the FDA-authorized assay, the prevalence of COVID-19 in that particular community (positivity rate over the previous 7-10 days or cases per population), and the clinical and epidemiological context of the person who has been tested.
The evaluation of a diagnostic antigen test result should consider the length of time the patient has experienced symptoms. Generally, clinicians can rely upon a positive diagnostic antigen test result because the specificity of current FDA-authorized antigen tests is high in a person who has COVID-19 symptoms.
The sensitivity of current FDA-authorized antigen tests varies, and thus negative diagnostic testing results should be handled differently depending on the testing device and its stated performance characteristics. In most cases, negative antigen diagnostic test results are considered presumptive. CDC recommends confirming negative antigen test results with an RT-PCR test when the pretest probability is relatively high, especially if the patient is symptomatic or has a known exposure to a person confirmed to have COVID-19. Ideally, confirmatory RT-PCR testing should take place within two days of the initial antigen testing. If RT-PCR testing is not available, clinical discretion can be used in whether to recommend the patient isolate. See CDC’s guidance on Quarantine and Isolation, Discontinuation of Isolation for Persons with COVID-19 Not in Healthcare Settings, Discontinuation of Transmission-Based Precautions of Patients in Healthcare Settings, and Return to Work for Healthcare Personnel. CDC does not recommend using antigen tests to make decisions about discontinuing isolation.
Currently, the rapid antigen tests that have received EUAs from FDA are authorized for diagnostic testing on symptomatic persons within the first five to seven days of symptom onset. See FDA’s In Vitro Diagnostics EUAs in addition to information from CMS on enforcement discretion for the use of SARS-CoV-2 point-of-care antigen diagnostic testing on asymptomatic individuals. Serial antigen testing within a closed congregate setting, such as a long-term care facility or a correctional facility, could quickly identify someone with a SARS-CoV-2 infection and prevent further transmission. Modeling evidence shows that outbreak control depends largely on the frequency of testing and the speed of reporting and is only marginally improved by high test sensitivity. For this reason, serial antigen testing may have benefits for early identification and controlling outbreaks in some situations, such as congregate living, compared to RT-PCR tests in settings with prolonged turnaround times. See FDA’s FAQ on use of antigen tests in high risk congregate settings.
When used for screening testing in congregate settings, test results for SARS-CoV-2 should be considered presumptive. Confirmatory nucleic acid testing following a positive antigen test may not be necessary when the pretest probability is high, especially if the person is symptomatic or has a known exposure. When the pretest probability is low, those persons who receive a positive antigen test should isolate until they can be confirmed by RT-PCR. See CDC’s Discontinuation of Isolation for Persons with COVID-19 Not in Healthcare Settings, Discontinuation of Transmission-Based Precautions of Patients in Healthcare Settings, and Return to Work for Healthcare Personnel.
Confirmatory nucleic acid testing following a negative antigen test used for screening testing may not be necessary if the pretest probability is low, the person is asymptomatic, or has no known exposures, or is part of a cohort that will receive rapid antigen tests on a recurring basis. Nucleic acid testing is also considered presumptive when screening asymptomatic persons, the potential benefits of confirmatory testing should be carefully considered in the context of person’s clinical presentation.
CDC will update this guidance as more data become available.
Reporting Rapid Antigen Test Results for SARS-CoV-2 to Health Departments and Patients
A CLIA-certified laboratory or testing site must report rapid antigen diagnostic test results to the local, state, tribal, or territory health department in accordance with Public Law 116-136, § 18115(a), the Coronavirus Aid, Relief, and Economic Security (CARES) Act, which requires “every laboratory that performs or analyzes a test that is intended to detect SARS-CoV-2 or to diagnose a possible case of COVID-19” to report the results of each such test. Antigen test results that are reported to public health departments must be clearly distinguished from other COVID-19 tests, such as RT-PCR tests and antibody tests.
On June 4, 2020, the US Department of Health and Human Services published guidance on COVID-19 Pandemic Response, Laboratory Data Reporting: CARES Act Section 18115 that specifies what additional data should be collected and electronically reported to health departments along with COVID-19 diagnostic or screening test results. Laboratory and testing professionals should collect and report complete patient demographic information and ensure that they report antigen test results using the proper LOINC code for their particular FDA-authorized assay(s). Facilities should refer to CDC’s LOINC In Vitro Diagnostic (LIVD) Test Code Mapping for SARS-CoV-2 Tests.
A CLIA-certified laboratory or testing site must report antigen test results to the individual or the individual’s healthcare provider according to the instructions for use of the FDA-authorized SARS-CoV-2 in vitro diagnostic device that was used. Depending on the stipulations of the FDA authorization, the laboratory or testing site may be required to report negative test results to patients as “presumptive negative.”
Laboratories that are either CLIA-certified or non CLIA-certified may conduct surveillance testing. Results of surveillance testing that uses rapid antigen tests can be returned in aggregate to the requesting institution, such as a university or public health agency. Negative antigen surveillance test results should be reported as “presumptive negative” to the requesting institution. Laboratories, regardless of their CLIA status, should not officially report the results of surveillance testing to health departments. If a local, state, tribal, or territory health department requests access to the results of surveillance testing for SARS-CoV-2 that uses antigen testing, the laboratory should state in the report to the health department that the data are antigen surveillance testing results that do not represent COVID-19 diagnostic or screening test results.
Laboratories that conduct surveillance testing, including surveillance testing that uses antigen tests, should not report test results to persons whose specimens have been tested, or to their health-care provider, employer, etc. Surveillance testing is performed on de-identified specimens, and thus results are not linked to an individual person. If at any time a laboratory intends to report a patient-specific test result, it must first obtain a CLIA certificate and meet all requirements to perform testing. For more information, see CMS’s summary of the CLIA regulations. Table 1 also summarizes the reporting requirements depending on the type of testing that is performed.